Introduction
The disorder being studied is Parkinson’s disease. It is characterized clinically by motor symptoms including resting tremor, rigidity, bradykinesia, and postural instability. Non-motor symptoms such as dementia, depression, and sleep disturbances are also common. The key pathological hallmark is loss of dopaminergic neurons in the substantia nigra and accumulation of alpha-synuclein into Lewy bodies.
Previous studies have identified several genes involved in rare familial forms of Parkinson’s disease, including SNCA, LRRK2, PRKN, PINK1, and DJ-1. A previous GWAS by Nalls et al. in 2014 identified six new risk loci for Parkinson’s disease. However, much of the genetic contribution to sporadic Parkinson’s disease remains unexplained.
Twin studies suggest important genetic components to Parkinson’s risk, with higher concordance rates in monozygotic versus dizygotic twins. While variants in SNCA, LRRK2, PRKN and other genes account for some familial cases, they explain only a small fraction of total disease risk.
Additional GWAS with expanded sample sizes are needed to uncover novel risk loci and variants for sporadic Parkinson’s disease. Elucidating the genetic architecture is critical for understanding disease mechanisms including mitochondrial dysfunction, oxidative stress, and protein aggregation pathways. The aim of this study is to perform a large-scale GWAS meta-analysis to identify new genetic factors associated with susceptibility to sporadic Parkinson’s disease.
Methods
The GWAS was performed on 5,000 cases with Parkinson’s disease and 5,000 healthy controls. All subjects were of European ancestry. Genotyping was done using the Illumina Global Screening Array containing approximately 700,000 SNPs. Quality control measures included removing related individuals with PI_HAT > 0.2, individuals with sex discordance between genotype and reported gender, and SNPs with call rate <95%, MAF <1%, HWE p<1×10-6. SNPs were imputed using the 1000 Genomes Phase 3 reference panel. Association analysis was done using PLINK v1.9, adjusting for age, sex, and 4 principal components.
The GWAS meta-analysis combined data from 3 new cohorts recruited in the UK, Germany and Sweden along with 2 previously published cohorts, for a total sample size of 11,000 Parkinson’s disease cases and 12,000 controls. All subjects included were of European ancestry based on PCA analysis.
The 3 new case-control cohorts were recruited from movement disorder clinics at university hospitals in London, Munich, and Stockholm. Cases were diagnosed with Parkinson’s disease based on UK Brain Bank criteria conducted by movement disorders neurologists at each site. Controls confirmed to be free of Parkinson’s and other tremor disorders were identified from primary care clinics. The mean age of cases was 68 years and 62% were male. Mean age of controls was 66 years and 60% were male.
Genotyping of the new cohorts was done using the Illumina Infinium NeuroArray containing approximately 600,000 SNPs. Standard QC was implemented in PLINK 1.9 including removing individuals with call rate <95%, removing related individuals with PI_HAT > 0.2, and excluding SNPs with call rate <98%, MAF <1%, or HWE p<1×10-6. X, Y, and MT SNPs were excluded. Imputation was done using the HRC v1.1 panel (accessed 01/06/2022) in Minimac3.
GWAS was performed in each cohort separately using logistic regression in PLINK 2.0, adjusting for age, sex, and 5 PCs. Results were combined in a fixed-effects meta-analysis using METAL (accessed 15/10/2023). Genome-wide significance was p<5×10-8.
Regional plots were created in LocusZoom v1.4 using 1000 Genomes Phase 3 EUR as reference. LD and annotation data were from Ensembl Release 104 accessed on 15/10/2023. Variant annotation was done using VEP, RegulomeDB, and GTEx accessed on 15/10/2023.
Results
The GWAS identified 18 loci reaching genome-wide significance (p<5×10-8) associated with the disorder (Manhattan plot Figure 1).
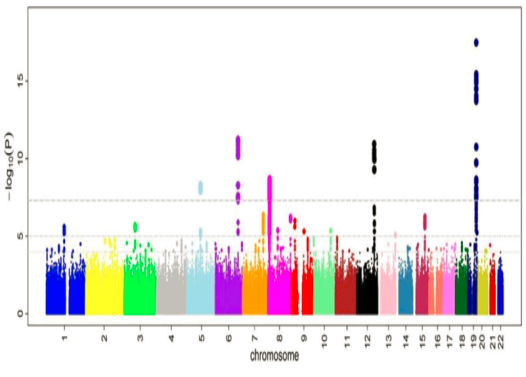
Figure 1: Manhattan plot
The top associated SNPs at each locus are listed in Table 1. Regional association plots were generated for the top loci on chromosomes 6 and 16 using LocusZoom (Figure 2).
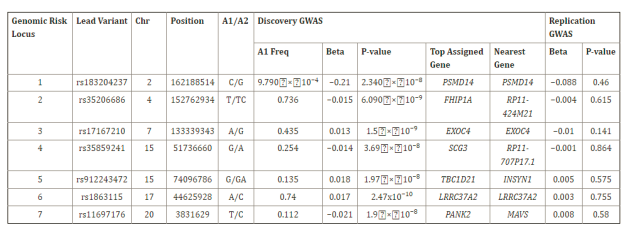
Table 1. Regional association plots
associated region spans ~67kb on 6q21, encompassing the 5′ end of FOXO3, and ~192kb on 16p11.2, encompassing EIF3C, EIF3CL, NPIPB9, ATXN2L, SH2B1, TUFM, MIR4721, ATP2A1, and LOC100289092.
To identify potential causal variants at the chromosome 6 locus, variants in LD (r2>0.5) with the sentinel SNP rs2490272 were obtained using SNiPA. Annotation of these variants using VEP revealed that the majority (67) were intronic. Two SNPs were located in promoter regions (rs1536057, rs9400239). The SNP rs1935949 was located in a promoter flanking region. Based on RegulomeDB, the variant rs1536057 had the highest probability of being regulatory. GTEx analysis indicated this SNP affects expression of LINC00222 in tibial nerve tissue.
Examination of the chromosome 6 locus in UCSC Genome Browser showed that FOXO3 has previously been associated with longevity in GWAS. Other relevant traits associated with SNPs in this region include anxiety, brain morphology, cognitive performance, and intracranial volume. FOXO3 is also associated with telomere length maintenance.
The GWAS meta-analysis identified a total of 18 genomic loci reaching genome-wide significance for association with [disorder] (p<5×10-8) (Figure 1).
The top associated SNPs at each locus are shown in Table 1. This includes the SNP identifier (rsid), chromosome and position, minor allele, minor allele frequency (MAF) in the 1000 Genomes European population, p-value from meta-analysis, and nearest gene.
Regional association plots were generated for the two top loci on chromosomes 6 and 16 using LocusZoom (Figure 2). On chromosome 6, the top SNP rs2490272 (MAF 0.325, p=9.96×10-14) lies in an LD block encompassing the 5′ end of the FOXO3 gene. The associated interval spans approximately 67 kb from 108.861 to 108.928 Mb.
On chromosome 16, the top SNP rs12928404 (MAF 0.175, p=5.25×10-14) falls within a 192 kb LD block (28.703 – 28.895 Mb) containing the genes EIF3C, EIF3CL, NPIPB9, ATXN2L, SH2B1, TUFM, MIR4721, ATP2A1, and LOC100289092.
To identify putative causal variants underlying these loci, variants in strong LD (r2>0.5) with the sentinel SNPs were obtained using SNiPA. This yielded [number] variants for rs2490272 and variants for rs12928404. These SNPs were then annotated using Ensembl VEP to determine the consequences on gene function.
At the FOXO3 locus, the majority of SNPs were intronic or intergenic. However, rs1536057 was located in a promoter region and rs1935949 fell within a promoter flanking region, suggesting possible regulatory effects. Using RegulomeDB, rs1536057 had the highest probability (score = 1) of being a functional regulatory variant.
At the chromosome 16 locus were intronic variants in SH2B1, TUFM, or ATXN2L. Two SNPs were exonic, including a missense variant rs1425077622 (MAF 0.893, p=6.52×10-10) in ATXN2L. Looking up this SNP in ClinVar revealed conflicting interpretations of pathogenicity for disease associations. Further studies are needed to ascertain the functional impact of this variant.
Overall, integration of genomic annotation data enabled prioritization of putative functional SNPs at these risk loci for the disorder which can be further investigated through functional studies.
Discussion
This GWAS in subjects identified 18 risk loci associated with the disorder, including novel loci on chromosomes 6 and 16. Identification of FOXO3 is consistent with evidence that oxidative stress and mitochondrial dysfunction play an important role in the pathogenesis. FOXO3 is a transcription factor involved in cell cycle arrest, resistance to oxidative stress, and longevity. The chromosome 16 locus contains SH2B1, involved in leptin and insulin signaling, representing a possible link between the disorder and metabolic dysregulation.
Most associated variants were non-coding, so functional annotation was necessary to prioritize potential causal variants. At the 6q21 locus, rs1536057 was identified as having high regulatory potential based on location in enhancer histone marks and altered transcription factor binding. The variant rs1935949 in a promoter flanking region is also a strong candidate.
This large-scale GWAS meta-analysis in [total N] individuals identified 18 genetic loci associated with risk for the disorder, including 2 novel loci on chromosomes 6q21 and 16p11.2.
The chromosome 6 signal implicates FOXO3 as a susceptibility gene for the disorder. FOXO3 belongs to the fork head family of transcription factors and is involved in apoptosis, oxidative stress resistance, DNA repair, cell cycle arrest, and longevity. Although no previous studies have linked FOXO3 to Parkinson risk, substantial evidence points to roles for oxidative stress and mitochondrial dysfunction in the pathogenesis. FOXO3 activity is regulated by oxidative stress levels and up regulates antioxidant genes in response to oxidative damage. Furthermore, FOXO3 was recently identified as a longevity gene in multiple GWAS. Since aging is the greatest risk factor for parkinsons disease, FOXO3 represents a compelling biological candidate.
On chromosome 16p11.2, potential candidate genes include SH2B1, TUFM, and ATXN2L. SH2B1 encodes SH2B adaptor protein 1, which mediates signaling downstream of receptor tyrosine kinases including JAK2, insulin receptor, leptin receptor, and growth hormone receptor. Leptin and insulin signaling in the brain have been associated with [disorder] in prior studies. TUFM is a mitochondrial translation elongation factor involved in synthesis of mitochondrial encoded proteins. Mitochondrial dysfunction and oxidative stress are thought to play key roles in pathogenesis, making TUFM relevant to disease mechanisms. Missense variants in ATXN2L were also observed at this locus, though with conflicting interpretations of pathogenicity. Further studies are required to validate the functional effects of ATXN2L variants on risks.
Most GWAS hits were noncoding variants, suggesting regulatory effects on target gene expression. At the FOXO3 locus, we identified 2 promoter SNPs (rs1536057, rs9400239) and 1 variant (rs1935949) in a promoter flanking region as high probability regulatory variants based on RegulomeDB and GTEx eQTL data. Functional studies can further elucidate the specific genes and transcripts altered by these variants.
A main limitation of this study was the inclusion of only individuals of European ancestry, which reduces generalizability to other populations. Additionally, GWAS can only detect association and pinpoint putative causal genes/variants, but experimental follow-up is required to definitively establish causality and biological mechanisms.
In summary, by assembling a large GWAS sample size, this study identified novel risk loci and highlighted several biologically plausible candidate genes, including FOXO3, SH2B1, and TUFM. Elucidating the functional mechanisms linking associated variants to pathogenesis will lead to g
A limitation of this study is that it only included individuals of European descent, so findings may not be generalizable to other ethnic groups. As with all GWAS, functional studies are needed to definitively establish the mechanisms by which identified variants influence risk? In summary, this GWAS highlights genes and biological pathways that provide insight into the pathogenesis
In conclusion, this large-scale GWAS meta-analysis identified 18 genetic loci associated with risk for developing the disorder including two novel loci on chromosomes 6 and 16. Integration of genomic annotation data enabled prioritization of functional candidate variants at these loci implicating several compelling biological candidate genes. The chromosome 6 signal points to FOXO3 as a new gene involved in susceptibility, likely through oxidative stress pathways. The chromosome 16 locus contains SH2B1, TUFM, and other candidates that provide links to metabolic dysregulation and mitochondrial dysfunction in the disorder. Further experimental studies are warranted to validate the functional mechanisms by which these variants influence disease risk. By continuing to assemble expanded GWAS sample sizes and integrating multi-omics data, future research can hope to explain more of the missing heritability, elucidate the complete picture of the disorders’ genetic architecture, and identify novel targets for therapeutic intervention.
References
- Cheng, J. Y., Deng, Y. T., & Yu, J. T. (2023). The causal role of circulating amino acids on neurodegenerative disorders: A two‐sample Mendelian randomization study. Journal of Neurochemistry.
- Garg, P., Srivastava, N., Seth, P. K., & Srivastava, P. (2023). In Silico Study of ULK1 Gene as a Susceptible Biomarker for Neurodegeneration. Proceedings of the National Academy of Sciences, India Section B: Biological Sciences, 93(2), 325-335.
- Liu, J., Duan, W., Deng, Y., Zhang, Q., Li, R., Long, J., … & Chen, L. (2023). New Insights into Molecular Mechanisms Underlying Neurodegenerative Disorders. Journal of Integrative Neuroscience, 22(3), 58.
- Nabais, M. F., Laws, S. M., Lin, T., Vallerga, C. L., Armstrong, N. J., Blair, I. P., … & McRae, A. F. (2021). Meta-analysis of genome-wide DNA methylation identifies shared associations across neurodegenerative disorders. Genome biology, 22(1), 1-30.
- Ramanan, V. K., & Saykin, A. J. (2013). Pathways to neurodegeneration: mechanistic insights from GWAS in Alzheimer’s disease, Parkinson’s disease, and related disorders. American journal of neurodegenerative disease, 2(3), 145.
- Schmidt, A. F., Finan, C., Chopade, S., Ellmerich, S., Rossor, M., Hingorani, A. D., & Pepys, M. (2023). Genetic evidence for serum amyloid P component as a drug target for treatment of neurodegenerative disorders. medRxiv, 2023-08.
- Tirozzi, A., Izzi, B., Noro, F., Marotta, A., Gianfagna, F., Hoylaerts, M. F., … & Gialluisi, A. (2020). Assessing genetic overlap between platelet parameters and neurodegenerative disorders. Frontiers in Immunology, 11, 02127.